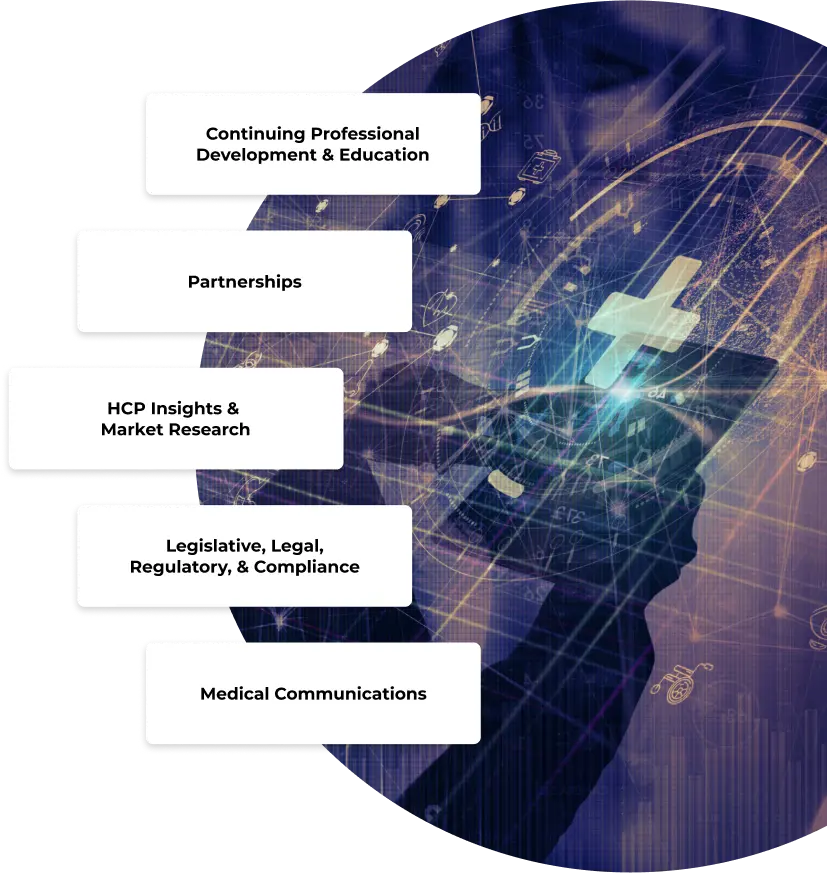
WHO WE ARE
Clinical Education Alliance (CEA) is a global leader in healthcare professional (HCP) engagement and education
At the heart of our alliance lies a unified commitment to enrich and improve the lives of over 1 billion patients.
Spanning across continents, disciplines, and specialties, we empower HCPs with cutting-edge healthcare education, meaningful insights, and innovative tools to help them stay current for improved patient outcomes.
Together, we pave the way for a brighter and healthier future for all.
Our Alliance




Network and Data
Science Backbone
Healthcare Professional
Engagement and Education